Après la FDA aux USA en février dernier, c’était au tour de l’AFSSAPS dans le courant de l’été de nous diffuser un courrier d’alerte concernant les risques de l’utilisation du Kenacort en intra-vitréen, et rappelant que cette utilisation, n’ayant fait l’objet d’aucune étude, est « hors AMM » :
Le laboratoire Bristol-Myers Squibb (BMS), en accord avec l’Agence Française de Sécurité Sanitaire des Produits de Santé (Afssaps) souhaite vous communiquer d’importantes données de pharmacovigilance concernant les spécialités KENACORT RETARD 40 mg/1ml et 80mg/2ml (suspensions injectables d’acétonide de triamcinolone):
Des cas graves d’endophtalmie, d’inflammation oculaire, d’augmentation de la pression intraoculaire et de troubles visuels incluant des cas de cécité, ont été rapportés à la suite d’administrations intravitréennes de KENACORT RETARD au cours d’utilisations hors-AMM. La plupart de ces cas ont nécessité une prise en charge thérapeutique ou chirurgicale.
A ce jour, aucune étude menée par BMS n’a évalué la tolérance de l’administration de KENACORT RETARD par injection sous-conjonctivale, sous-ténonienne, rétrobulbaire ou intraoculaire (voie intravitréenne).
Ces constatations sont elles suffisantes pour avoir peur de continuer à utiliser le Kenacort ? Probablement non.
- Si BMS n’a effectivement fait aucune étude concernant le Kenacort utilisé en intra-vitréen, les publications sur le sujet ne manquent pas, surtout depuis 2003 :
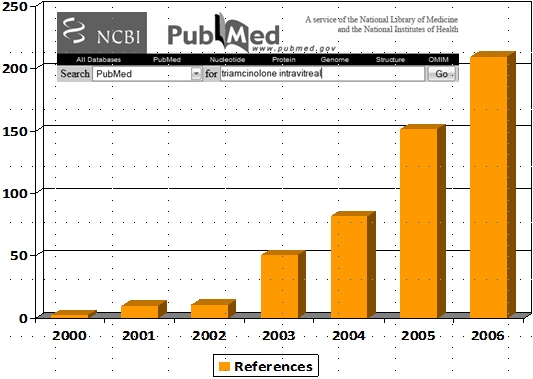
- Les effets secondaires rapportés n’ont finalement pas grand chose d’inquiétant :
- une endophtalmie peut survenir à la suite de toute injection intravitréenne : leur existence n’a pas empêché la mise sur le marché des anti-VEGF en traitement de la DMLA (avec des taux dans certaines études parfois plus importants que ceux actuellement rapportés). De même, ce risque ne nous empêche pas d’opérer des cataractes (chez des patients avec parfois des niveaux de vision préopératoires bien supérieurs à ceux à qui nous proposons le Kenacort…);
- Une hypertonie peut survenir après tout traitement corticoïde, quelle qu’en soit la modalité d’administration (on parle de près de 30% de « répondeurs » après traitement par collyres corticoïdes, et cela ne nous empêche pas de les utiliser largement).
Si l’un des corticoïdes actuellement en développement pour un usage spécifique en ophtalmologie (j’en reparle bientôt) arrive sur le marché, ce sera probablement avec le même type d’effets secondaires…
A mon avis, cette lettre nous rappelle donc juste notre devoir : bien évaluer le rapport bénéfice-risque avant de poser une indication, raisonnée (ce qui est finalement relativement facile en l’absence d’alternative !) et en informer nos patients (comme avant toute procédure).
Par contre, plus que les complications sus-citées, c’est peut-être une éventuelle toxicité du Kenacort (pour le moment encore controversée : des études animales rapportent régulièrement l’existence de phénomènes toxiques des conservateurs et/ou de la triamcinolone elle-même, tandis qu’à ma connaissance, aucune série clinique n’est venue confirmer ces données) qui pourrait nous faire changer d’attitude.
Quoi qu’il en soit, vivement qu’arrivent les premiers résultats des grandes études randomisées actuellement menées aux USA sous l’égide du National Eye Institute (en traitement de l‘oedème maculaire diabétique et des occlusions veineuses)… avec la réserve que le produit utilisé est une triamcinolone spécialement fabriquée par Allergan, justement sans conservateur…